12.1 Mechanism of Action
The symptoms associated with benign prostatic hyperplasia (BPH) are related to bladder outlet obstruction, which is comprised of two underlying components: static and dynamic. The static component is related to an increase in prostate size caused, in part, by a proliferation of smooth muscle cells in the prostatic stroma. However, the severity of BPH symptoms and the degree of urethral obstruction do not correlate well with the size of the prostate. The dynamic component is a function of an increase in smooth muscle tone in the prostate and bladder neck leading to constriction of the bladder outlet. Smooth muscle tone is mediated by the sympathetic nervous stimulation of alpha
Step 2: Wait till a Gold Button appears, click on it.
Step 3: The “Claim My Reward” link will appear at the bottom of the article, scroll down and click on it.
1 adrenoceptors, which are abundant in the prostate, prostatic capsule, prostatic urethra, and bladder neck. Blockade of these adrenoceptors can cause smooth muscles in the bladder neck and prostate to relax, resulting in an improvement in urine flow rate and a reduction in symptoms of BPH.
Tamsulosin, an alpha
1 adrenoceptor blocking agent, exhibits selectivity for alpha
1 receptors in the human prostate. At least three discrete alpha
1 adrenoceptor subtypes have been identified: alpha
1A, alpha
1B, and alpha
DOUBLE REWARD!
Watch the Video Till The End!
1D; their distribution differs between human organs and tissue. Approximately 70% of the alpha
1 receptors in human prostate are of the alpha
1A subtype.
Tamsulosin hydrochloride capsules are not intended for use as an antihypertensive drug.
12.2 Pharmacodynamics
Urologic pharmacodynamic effects have been evaluated in neurologically impaired pediatric patients and in adults with BPH [see
USE IN SPECIFIC POPULATIONS (
8.4)
and
CLINICAL STUDIES (
14)
].
12.3 Pharmacokinetics
The pharmacokinetics of tamsulosin hydrochloride have been evaluated in adult healthy volunteers and patients with BPH after single and/or multiple administration with doses ranging from 0.1 mg to 1 mg.
Absorption
Absorption of tamsulosin hydrochloride from tamsulosin hydrochloride capsules 0.4 mg is essentially complete (>90%) following oral administration under fasting conditions. Tamsulosin hydrochloride exhibits linear kinetics following single and multiple dosing, with achievement of steady-state concentrations by the fifth day of once-a-day dosing.
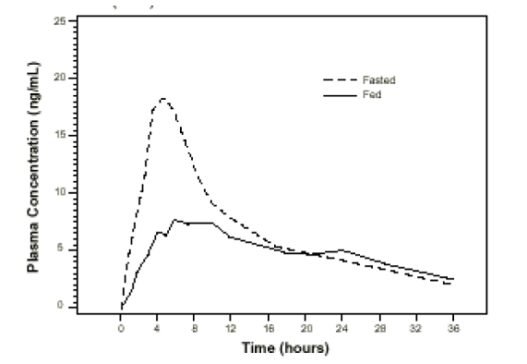
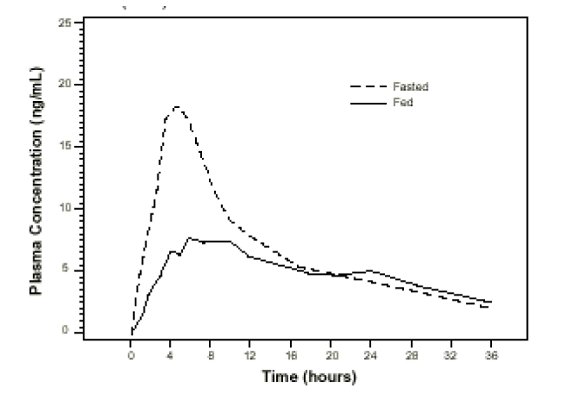
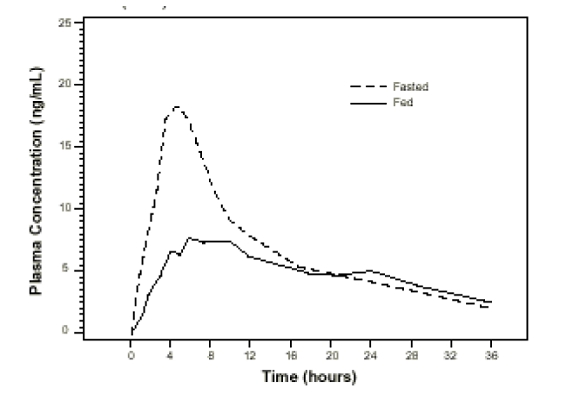
Effect of Food
The time to maximum concentration (T
max) is reached by 4 to 5 hours under fasting conditions and by 6 to 7 hours when tamsulosin hydrochloride capsules are administered with food. Taking tamsulosin hydrochloride capsules under fasted conditions results in a 30% increase in bioavailability (AUC) and 40% to 70% increase in peak concentrations (C
max) compared to fed conditions (
Figure 1).
Figure 1 Mean Plasma Tamsulosin Hydrochloride Concentrations Following Single-Dose Administration of Tamsulosin Hydrochloride Capsules, 0.4 mg Under Fasted and Fed conditions (n=8)
The effects of food on the pharmacokinetics of tamsulosin hydrochloride are consistent regardless of whether a tamsulosin hydrochloride capsule is taken with a light breakfast or a high-fat breakfast (
Table 2).
- Table 2 Mean (± S.D.) Pharmacokinetic Parameters Following Tamsulosin Hydrochloride Capsules 0.4 mg Once Daily or 0.8 mg Once Daily with a Light Breakfast, High-Fat Breakfast or Fasted
C
min = observed minimum concentration
C
max = observed maximum tamsulosin hydrochloride plasma concentration
T
max = median time-to-maximum concentration
T
1/2 = observed half-life
AUC
τ = area under the tamsulosin hydrochloride plasma time curve over the dosing interval
Distribution
The mean steady-state apparent volume of distribution of tamsulosin hydrochloride after intravenous administration to 10 healthy male adults was 16 L, which is suggestive of distribution into extracellular fluids in the body.
Tamsulosin hydrochloride is extensively bound to human plasma proteins (94% to 99%), primarily alpha
1 acid glycoprotein (AAG), with linear binding over a wide concentration range (20 to 600 ng/mL). The results of two-way
in vitro studies indicate that the binding of tamsulosin hydrochloride to human plasma proteins is not affected by amitriptyline, diclofenac, glyburide, simvastatin plus simvastatin-hydroxy acid metabolite, warfarin, diazepam, propranolol, trichlormethiazide, or chlormadinone. Likewise, tamsulosin hydrochloride had no effect on the extent of binding of these drugs.
Metabolism
There is no enantiomeric bioconversion from tamsulosin hydrochloride [R(-) isomer] to the S(+) isomer in humans. Tamsulosin hydrochloride is extensively metabolized by cytochrome P450 enzymes in the liver and less than 10% of the dose is excreted in urine unchanged. However, the pharmacokinetic profile of the metabolites in humans has not been established. Tamsulosin is extensively metabolized, mainly by CYP3A4 and CYP2D6, as well as via some minor participation of other CYP isoenzymes. Inhibition of hepatic drug-metabolizing enzymes may lead to increased exposure to tamsulosin
[see
WARNINGS AND PRECAUTIONS(
5.2)
and
DRUG INTERACTIONS (
7.1)
]. The metabolites of tamsulosin hydrochloride undergo extensive conjugation to glucuronide or sulfate prior to renal excretion.
Incubations with human liver microsomes showed no evidence of clinically significant metabolic interactions between tamsulosin hydrochloride and amitriptyline, albuterol (beta agonist), glyburide (glibenclamide) and finasteride (5alpha-reductase inhibitor for treatment of BPH). However, results of the
in vitro testing of the tamsulosin hydrochloride interaction with diclofenac and warfarin were equivocal.
Excretion
On administration of the radiolabeled dose of tamsulosin hydrochloride to 4 healthy volunteers, 97% of the administered radioactivity was recovered, with urine (76%) representing the primary route of excretion compared to feces (21%) over 168 hours.
Following intravenous or oral administration of an immediate-release formulation, the elimination half-life of tamsulosin hydrochloride in plasma ranged from 5 to 7 hours. Because of absorption rate-controlled pharmacokinetics with tamsulosin hydrochloride capsules, the apparent half-life of tamsulosin hydrochloride is approximately 9 to 13 hours in healthy volunteers and 14 to 15 hours in the target population.
Tamsulosin hydrochloride undergoes restrictive clearance in humans, with a relatively low systemic clearance (2.88 L/h).
Specific Populations
Pediatric Use
Tamsulosin hydrochloride capsules are not indicated for use in pediatric populations [see
USE IN SPECIFIC POPULATIONS (
8.4)
].
Geriatric (Age) Use
Cross-study comparison of tamsulosin hydrochloride capsules overall exposure (AUC) and half-life indicates that the pharmacokinetic disposition of tamsulosin hydrochloride may be slightly prolonged in geriatric males compared to young, healthy male volunteers. Intrinsic clearance is independent of tamsulosin hydrochloride binding to AAG, but diminishes with age, resulting in a 40% overall higher exposure (AUC) in subjects of age 55 to 75 years compared to subjects of age 20 to 32 years [see
USE IN SPECIFIC POPULATIONS (
8.5)
].
Renal Impairment
The pharmacokinetics of tamsulosin hydrochloride have been compared in 6 subjects with mild-moderate (30≤CL
cr<70 mL/min/1.73 m
2) or moderate-severe (10≤CL
cr<30 mL/min/1.73 m
2) renal impairment and 6 normal subjects (CL
cr>90 mL/min/1.73 m
2). While a change in the overall plasma concentration of tamsulosin hydrochloride was observed as the result of altered binding to AAG, the unbound (active) concentration of tamsulosin hydrochloride, as well as the intrinsic clearance, remained relatively constant. Therefore, patients with renal impairment do not require an adjustment in tamsulosin hydrochloride capsules dosing. However, patients with endstage renal disease (CL
cr<10 mL/min/1.73 m
2) have not been studied [see
USE IN SPECIFIC POPULATIONS (
8.6)
].
Hepatic Impairment
The pharmacokinetics of tamsulosin hydrochloride have been compared in 8 subjects with moderate hepatic impairment (Child-Pugh’s classification: Grades A and B) and 8 normal subjects. While a change in the overall plasma concentration of tamsulosin hydrochloride was observed as the result of altered binding to AAG, the unbound (active) concentration of tamsulosin hydrochloride does not change significantly, with only a modest (32%) change in intrinsic clearance of unbound tamsulosin hydrochloride. Therefore, patients with moderate hepatic impairment do not require an adjustment in tamsulosin hydrochloride capsules dosage. Tamsulosin hydrochloride has not been studied in patients with severe hepatic impairment [see
USE IN SPECIFIC POPULATIONS(
8.7)
].
Drug Interactions
Cytochrome P450 Inhibition
Strong and Moderate Inhibitors of CYP3A4 or CYP2D6
The effects of ketoconazole (a strong inhibitor of CYP3A4) at 400 mg once daily for 5 days on the pharmacokinetics of a single tamsulosin hydrochloride capsule 0.4 mg dose was investigated in 24 healthy volunteers (age range 23 to 47 years). Concomitant treatment with ketoconazole resulted in an increase in the C
max and AUC of tamsulosin by a factor of 2.2 and 2.8, respectively [see
WARNINGS AND PRECAUTIONS(
5.2)
and
CLINICAL PHARMACOLOGY (
12.3)
]. The effects of concomitant administration of a moderate CYP3A4 inhibitor (e.g., erythromycin) on the pharmacokinetics of tamsulosin hydrochloride have not been evaluated [see
WARNINGS AND PRECAUTIONS (
5.2)
and
DRUG INTERACTIONS (
7.1)
].
The effects of paroxetine (a strong inhibitor of CYP2D6) at 20 mg once daily for 9 days on the pharmacokinetics of a single tamsulosin hydrochloride capsule 0.4 mg dose was investigated in 24 healthy volunteers (age range 23 to 47 years). Concomitant treatment with paroxetine resulted in an increase in the C
max and AUC of tamsulosin by a factor of 1.3 and 1.6, respectively [see
WARNINGS AND PRECAUTIONS(
5.2)
and
DRUG INTERACTIONS (
7.1)
]. A similar increase in exposure is expected in CYP2D6 poor metabolizers (PM) as compared to extensive metabolizers (EM). A fraction of the population (about 7% of Caucasians and 2% of African Americans) are CYP2D6 PMs. Since CYP2D6 PMs cannot be readily identified and the potential for significant increase in tamsulosin exposure exists when tamsulosin hydrochloride 0.4 mg is co-administered with strong CYP3A4 inhibitors in CYP2D6 PMs, tamsulosin hydrochloride 0.4 mg capsules should not be used in combination with strong inhibitors of CYP3A4 (e.g., ketoconazole) [see
WARNINGS AND PRECAUTIONS (
5.2)
and
DRUG INTERACTIONS (
7.1)
].
The effects of concomitant administration of a moderate CYP2D6 inhibitor (e.g., terbinafine) on the pharmacokinetics of tamsulosin hydrochloride have not been evaluated [see
WARNINGS AND PRECAUTIONS (
5.2)
and
DRUG INTERACTIONS(
7.1)
].
The effects of co-administration of both a CYP3A4 and a CYP2D6 inhibitor with tamsulosin hydrochloride capsules have not been evaluated. However, there is a potential for significant increase in tamsulosin exposure when tamsulosin hydrochloride 0.4 mg is co-administered with a combination of both CYP3A4 and CYP2D6 inhibitors [see
WARNINGS AND PRECAUTIONS (
5.2)
and
DRUG INTERACTIONS(
7.1)
].
Cimetidine
The effects of cimetidine at the highest recommended dose (400 mg every 6 hours for 6 days) on the pharmacokinetics of a single tamsulosin hydrochloride capsule 0.4 mg dose was investigated in 10 healthy volunteers (age range 21 to 38 years). Treatment with cimetidine resulted in a significant decrease (26%) in the clearance of tamsulosin hydrochloride, which resulted in a moderate increase in tamsulosin hydrochloride AUC (44%) [see
WARNINGS AND PRECAUTIONS(
5.2)
and
DRUG INTERACTIONS (
7.1)
].
Other Alpha Adrenergic Blocking Agents
The pharmacokinetic and pharmacodynamic interactions between tamsulosin hydrochloride capsules and other alpha adrenergic blocking agents have not been determined; however, interactions between tamsulosin hydrochloride capsules and other alpha adrenergic blocking agents may be expected [see
WARNINGS AND PRECAUTIONS (
5.2)
and
DRUG INTERACTIONS(
7.2)
].
PDE5 Inhibitors
Caution is advised when alpha adrenergic blocking agents, including tamsulosin hydrochloride, are co-administered with PDE5 inhibitors. Alpha-adrenergic blockers and PDE5 inhibitors are both vasodilators that can lower blood pressure. Concomitant use of these two drug classes can potentially cause symptomatic hypotension [see
WARNINGS AND PRECAUTIONS (
5.2)
and
DRUG INTERACTIONS (
7.3)
].
Warfarin
A definitive drug-drug interaction study between tamsulosin hydrochloride and warfarin was not conducted. Results from limited
in vitro and
in vivo studies are inconclusive. Therefore, caution should be exercised with concomitant administration of warfarin and tamsulosin hydrochloride capsules [see
WARNINGS AND PRECAUTIONS (
5.2)
and
DRUG INTERACTIONS (
7.4)
].
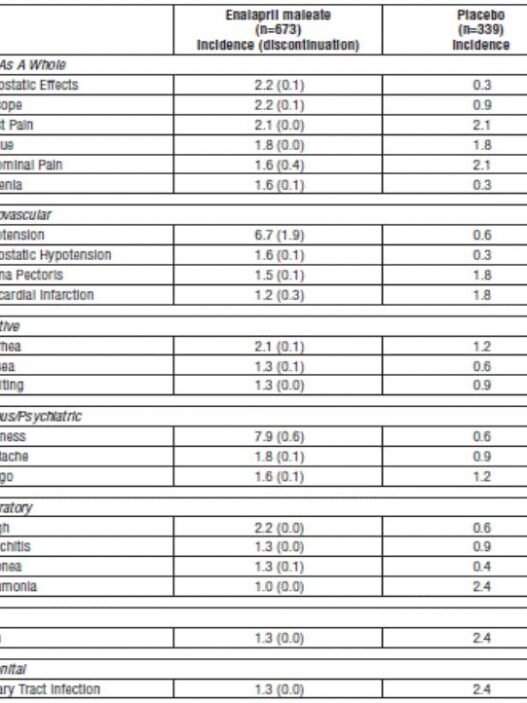
Nifedipine, Atenolol, Enalapril
In three studies in hypertensive subjects (age range 47 to 79 years) whose blood pressure was controlled with stable doses of nifedipine, atenolol, or enalapril for at least 3 months, tamsulosin hydrochloride capsules 0.4 mg for 7 days followed by tamsulosin hydrochloride capsules 0.8 mg for another 7 days (n=8 per study) resulted in no clinically significant effects on blood pressure and pulse rate compared to placebo (n=4 per study). Therefore, dosage adjustments are not necessary when tamsulosin hydrochloride capsules are administered concomitantly with nifedipine, atenolol, or enalapril [see
DRUG INTERACTIONS(
7.5)
].
Digoxin and Theophylline
In two studies in healthy volunteers (n=10 per study; age range 19 to 39 years) receiving tamsulosin hydrochloride capsules 0.4 mg/day for 2 days, followed by tamsulosin hydrochloride capsules 0.8 mg/day for 5 to 8 days, single intravenous doses of digoxin 0.5 mg or theophylline 5 mg/kg resulted in no change in the pharmacokinetics of digoxin or theophylline. Therefore, dosage adjustments are not necessary when a tamsulosin hydrochloride capsule is administered concomitantly with digoxin or theophylline [see
DRUG INTERACTIONS (
7.6)
].
Furosemide
The pharmacokinetic and pharmacodynamic interaction between tamsulosin hydrochloride capsules 0.8 mg/day (steady-state) and furosemide 20 mg intravenously (single dose) was evaluated in 10 healthy volunteers (age range 21 to 40 years). Tamsulosin hydrochloride capsules had no effect on the pharmacodynamics (excretion of electrolytes) of furosemide. While furosemide produced an 11% to 12% reduction in tamsulosin hydrochloride C
max and AUC, these changes are expected to be clinically insignificant and do not require adjustment of the tamsulosin hydrochloride capsules dosage [
see DRUG INTERACTIONS (
7.7)
].
Next Article